Scientists and engineers are on a quest to develop electronic devices that are compatible with our bodies: think of materials that can help wire neurons back together after brain injuries, or diagnostic tools that can easily be absorbed within the body.
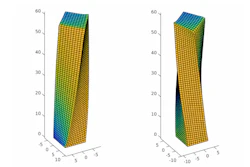
A family of self-assembling peptides, called π-conjugated oligopeptides, has shown promise for becoming the basis of the next-generation of these electronic, biocompatible materials. But identifying the right molecular sequences to create the optimal self-assembled nanostructures would require testing thousands of possibilities that each take approximately one month to test in the lab.
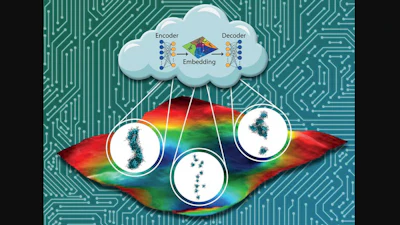
Assoc. Prof. Andrew Ferguson and his collaborators have sped up that process by developing machine learning tools that can screen for the best candidates. By screening 8,000 candidates of self-assembled peptides, the team was able to rank each design. That paves the way for experimentalists to test the most promising candidates.
The results were published in the journal J. Phys. Chem. B. The paper was also selected as the ACS Editors' Choice, which offers free public access to new research of importance to the global scientific community, and to be featured on the journal cover.
"By understanding data science, materials science, and molecular science, we were able to find an innovative way to screen for new possible candidates," Ferguson said. "The fact that this paper was chosen as an ACS Editors' Choice shows that there is a lot of interest in coupling artificial intelligence to domain science. It's an important problem that is of broad interest to the physical chemistry community."
Ranking peptides for experimentalists
To help find the best candidates, Ferguson and graduate student Kirill Shmilovich screened a family of π-conjugated oligopeptides using machine learning and molecular simulation. The set included 8,000 potential peptides, if researchers kept the same core and just changed the three amino acids on each side of the molecule. (The amino acids on the sides are symmetrical -- if you change one on one side, it changes on the other side, as well.)
Using a form of machine learning known as active learning or Bayesian optimization to guide molecular simulations, they were able to construct reliable data-driven models of how the sequence of the peptide influenced its properties after considering only 186 peptides.
The model predictions could then be reliably extrapolated to predict the properties of the rest of the peptide family. The process also removed human bias from the equation, letting artificial intelligence find features of peptide designs that researchers hadn't considered before that actually made them better candidates.
They then ranked each peptide and handed off their results to their experimental collaborators, who will then test the top candidates in the lab. Next, they hope to expand their system to include trying out different π-conjugated cores, while feeding new experimental data back into the loop to further strengthen their models.
They also hope to use this machine learning system for designing proteins, optimizing self-assembling colloids to make atomic crystals, and even to one day incorporate these tools into a self-driving laboratory, where artificial intelligence would take data, create predictions, run experiments, then feed that data back to the model -- all without human intervention.
"This is a method that could be useful in many different domains," Ferguson said.